임상시험에서는 원하는 결과를 얻기 위하여
엄격한 선정/제외 기준에 따라 적합한 대상자를 선정한다.
그러나 의약품 허가 후 실제 필드에서 사용될 경우에는
남녀노소 구분 없이 다양한 사람들이 복용을 하게 되고
복용기간 또한 일정하지 않아,
임상시험에서 발견되지 않았던
다양한 이상사례가 발생할 가능성이 높을 수 밖에 없다.
이에 국내에서는 품목허가를 받고 시판되는 의약품의
유효성과 안전성을 일정기간 동안 관찰하여
다시 한 번 의약품을 재심사하는 과정을 통해
의약품의 허가사항을 관리한다.
♣ 재심사 workflow

시판 최소 1개월 전까지 재심사 계획서를 작성하여
식약처에 제출하여야 한다.
재심사 기간 중 허가 후 2년까지는 매 6개월마다 정기보고를 하고,
2년 이후에는 1년마다 정기보고를 하며,
마지막 정기보고는 ‘재심사 신청’으로 갈음하는데,
재심사신청은 재심사 기간 만료 후 3개월 이내에 해야 한다.
재심사 신청을하게되면
식약처의 각 심사부서에서 재심사보고서를 심사하고,
결과에 따라 허가사항 변경 등의 후속 조치가 따르게 된다.
♣ 재심사 기간
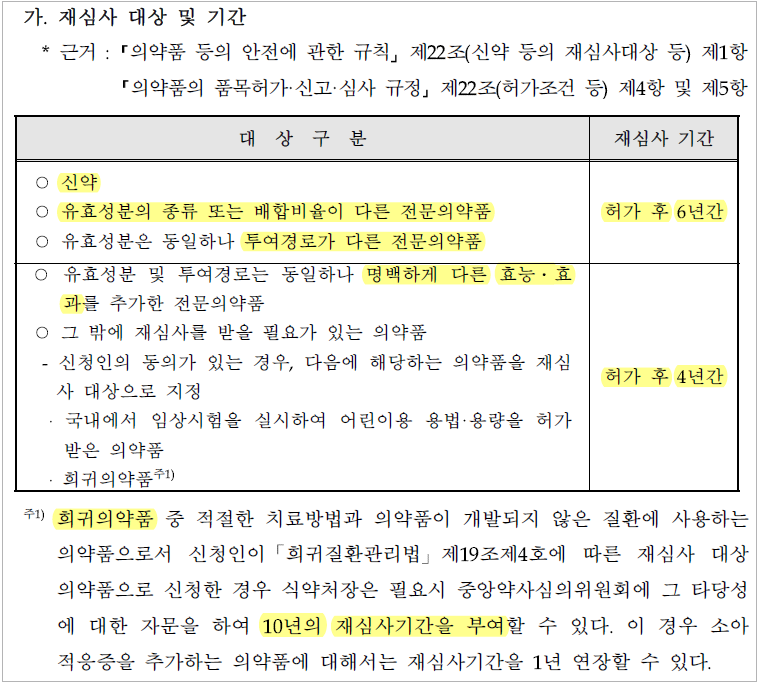
재심사 기간은 종류에 따라
허가일로부터 4년 이상 6년 이하로 지정되며,
희귀의약품의 경우 10년까지 가능하다.
♣ 조사대상자 수

의약품의 특성을 고려하여
해당 적응증의 국내 환자수, 유사품목의 처방건수,
급여청구액 등의 타당한 근거자료를 바탕으로
조사대상자수의 조절이 가능하다.
최근에는 위해성 관리계획(RMP, Risk Management Plan)
지정 의약품이 증가하며서
재심사의약품과의 자료가 중복되는 문제가 많아져,
식약처에서는 재심사제도와 위해성 관리계획으로
이원화되어있는 현재의 안전관리 제도를
단계적으로 위해성 관리계획으로 일원화하는 방안을
마련하여 추진할 계획이다.
하지만 한 번에 재심사를 없앨 수는 없으므로,
단계적으로 추진할 계획이라
당분간은 계속해서 재심사제도 또한
동시에 시행될 예정이다:)
'소담한 스터디' 카테고리의 다른 글
| 임상시험에서 눈가림(Blinding)의 종류 (0) | 2020.07.24 |
|---|---|
| 임상시험에서 약물이상반응의 예측성(Expectedness) 평가 (0) | 2020.07.24 |
| 희귀의약품이란?! (희귀의약품 지정 조건 및 지정 혜택) (0) | 2020.07.14 |
| 의약품 시판 전/시판 후 안전성 정보 보고 (0) | 2020.07.07 |
| 임상시험 안전성정보 보고 관련 규정 정리 (0) | 2020.07.06 |




댓글